INFORMACIÓN GENERAL
La miastenia grave (MG) es una enfermedad neuromuscular poco frecuente que se caracteriza porque los pacientes tienen fatigabilidad y debilidad de los músculos voluntarios.
La debilidad puede afectar cualquier músculo incluyendo músculos oculares, de las extremidades y músculos vitales, responsables de la función respiratoria, de la deglución y de la fonación .
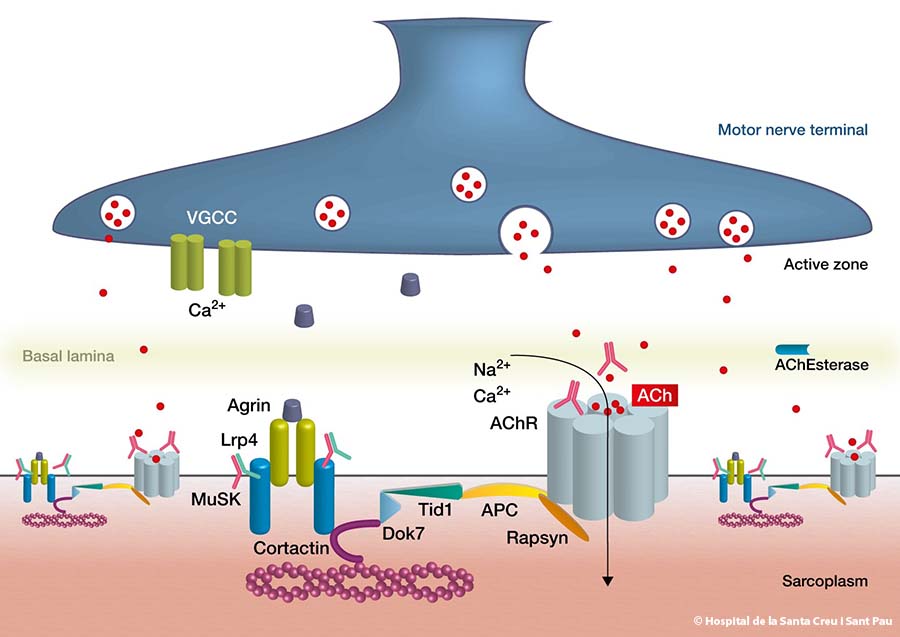
La enfermedad es autoinmune, es decir, se producen anticuerpos frente a proteínas propias localizadas en la membrana post-sináptica de la unión neuromuscular (la placa motora). Así pues, la enfermedad no se localiza en los nervios ni en los músculos propiamente sino en el sitio de unión entre nervio y músculo.
La MG es tratable y es por este motivo que debe ser reconocida y diagnosticada.
Idealmente, los pacientes deberían ser tratados por neurólogos expertos en Miastenia y seguidos y controlados por sus médicos de familia.
LA MG ES UNA ENFERMEDAD DE JÓVENES?
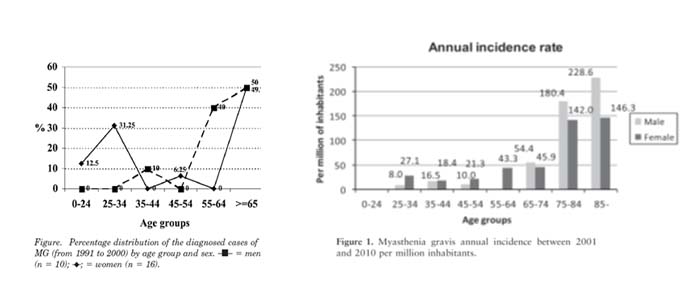
La MG se describía clásicamente como una enfermedad de pacientes jóvenes, entre 15-35 años y con un predominio de mujeres, pero actualmente se sabe que afecta predominantemente pacientes mayores de 60 años. En diferentes estudios, también en nuestro entorno (ver figura) se constata que más del 65% de los pacientes nuevos tienen más de 60 años. Este es un dato muy importante ya que tenemos una de las esperanzas de vida mayores del mundo. Se debe incluir la MG en el diagnóstico diferencial cuando la clínica sea compatible aunque el paciente sea muy mayor ya que es una enfermedad tratable con una muy buena respuesta, sobretodo en los ancianos.
QUÉ SE CONOCE SOBRE LOS ANTICUERPOS EN LA MG
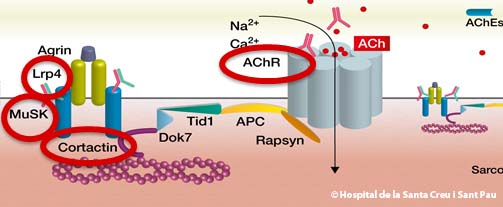
- En más del 80% de casos, La MG es secundaria a la presencia de anticuerpos frente al receptor de acetilcolina presente en la membrana post-sináptica de la unión neuromuscular: MG-RAch+
- Un porcentaje de los pacientes con MG-RAch negativos, tienen anticuerpos frente a otra molécula, MuSK, que está localizada en la misma región de la unión neuromuscular : MG-MuSK+
- En 2012, se describió un tercer antígeno, LRP4 en un pequeño número de enfermos, MG-LRP4+
- Nuestro grupo ha descrito en 2014 un nuevo antígeno, cortactina (CTTN), en un grupo de pacientes con MG ocular o generalizada IIA, MG-CTTN +
- El 5-7% restante son pacientes con una MG-“Seronegativa”, en la que todavía hoy no se ha demostrado ningún anticuerpo patogénico, aunque responden a los tratamientos inmunosupresores.
Así pués la MG es una enfermedad heterogénea no solo clínicamente sino inmunológicamente, con anticuerpos frente a como mínimo 4 antígenos (RAch, mayoritario, MuSK , LRP4, cortactin).
CUÁLES SON LAS MANIFESTACIONES CLÍNICAS
La principal queja clínica en la MG es la fatiga muscular que se acompaña de debilidad muscular. Empeora a lo largo del día y mejora total o parcialmente con el reposo. Los síntomas y signos de la miastenia dependen en todo momento de la musculatura implicada que incluye:
- Un 85-90 % de los pacientes debutan con afectación de la musculatura ocular , que produce diplopía (visión doble) y ptosis palpebral unilateral o bilateral, oscilante y no necesariamente siempre presente.
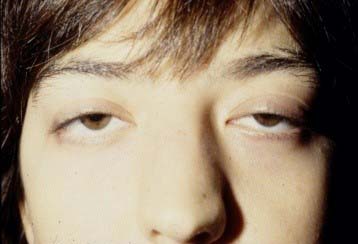
Ptosis palpebral bilateral - Debilidad de la musculatura facial, sobretodo del orbicular de los ojos y de los labios, que confiere una fascies característica.
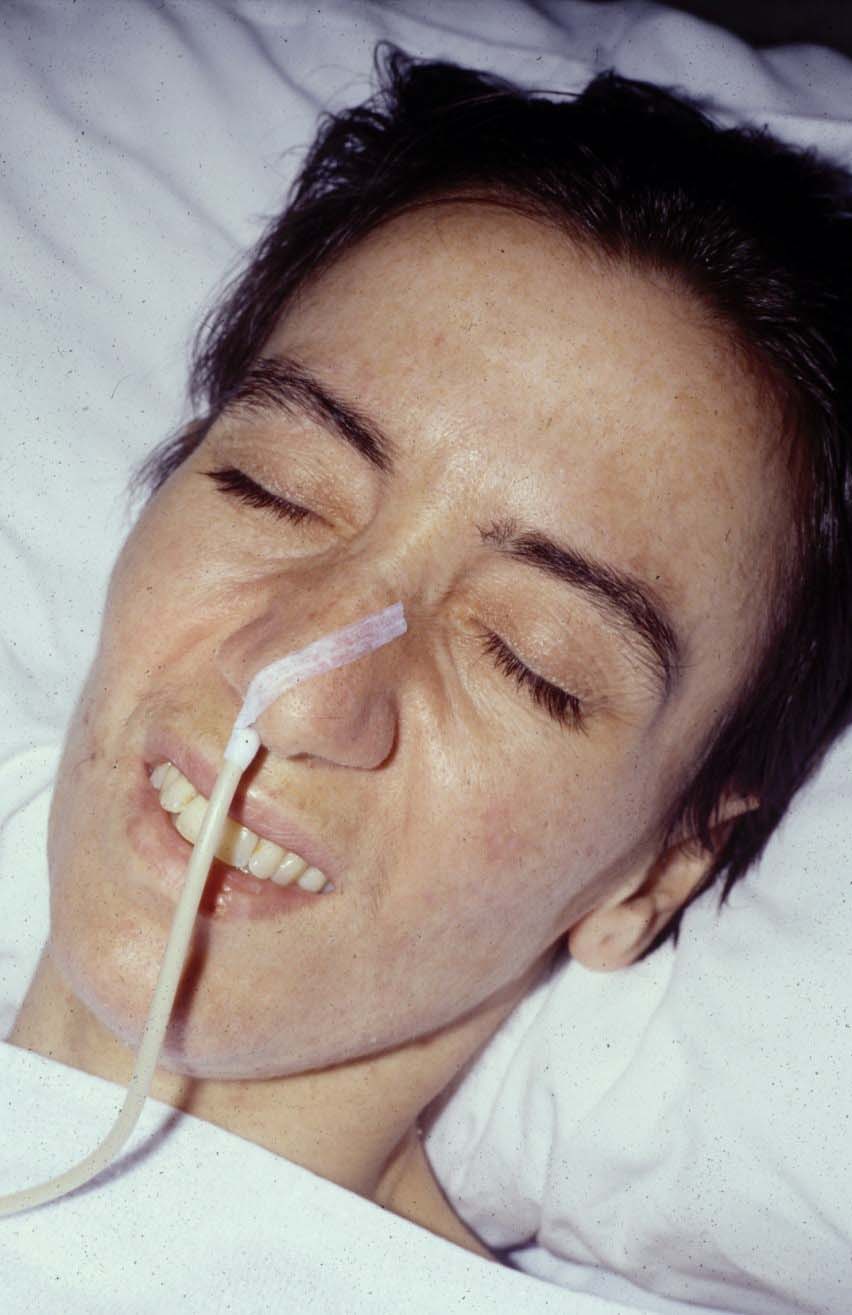
Debilidad facial orbicular ojos y de los labios - La afectación de la musculatura cervical provoca debilidad para la extensión del cuello y en numerosas ocasiones la de los músculos maseteros claudicación mandibular. Para demostrar la debilidad muscular con la fatiga, es de gran ayuda solicitar al paciente que , en decúbito prono, levante la cabeza hasta 20 veces (ver fotos en exploración)
- La afectación de la musculatura bulbar produce disfagia, disfonía, disartria y disnea. Es la más frecuente en los ancianos y debe ser reconocida porque es la que puede comprometer la vida de los pacientes. La clínica es oscilante y empeora cuando el paciente repite un movimiento.
Si la debilidad es importante no se puede objetivar en la historia ni en la exploración esta oscilación. Ante un enfermo con sospecha de MG y afectación bulbar con anticuerpos antiRAch negativos debe realizarse el estudio de los anticuerpos antiMuSK ya que estos están pueden ser positivos.
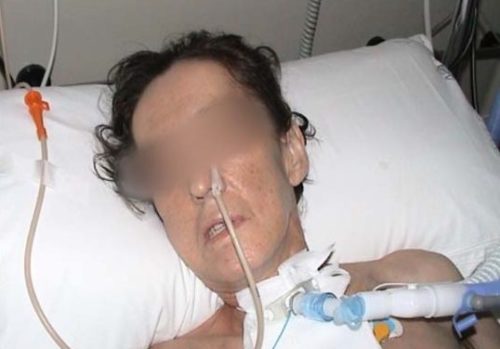
Una situación de urgencia es la crisis miasténica en la que se produce una insuficiencia respiratoria aguda; como consecuencia de la afectación de los músculos respiratorios. Requiere ingreso hospitalario y en algunas ocasiones la ventilación mecánica asistida.
- Debilidad y fatigabilidad de los músculos de las extremidades superiores e inferiores sobretodo a nivel proximal aunque se han descrito inicios de la enfermedad con debilidad exclusiva de los músculos distales. El paciente presenta generalmente dificultad para subir y bajar escaleras, manipular objetos por encima de su cabeza,…
Es importante plantear el diagnóstico de M.G no solo ante aquellos pacientes que refieran diplopia y ptosis palpebral sino también en aquellos enfermos, sobretodo viejos, que tienen disartria o disfagia que varía a lo largo del día. No es infrecuente que se les diagnostique de enfermedades mucho más prevalentes, como por ejemplo de patología vascular cerebral.
CLASIFICACIÓN CLÍNICA
En el año 2000 la Myasthenia Gravis Foundation of America (MGFA) propuso la clasificación que más se utiliza en este momento.
MG ocular:
- MG – I se reserva para los pacientes con debilidad solo de la musculatura ocular.
MG generalizada:
MG grados II, III y IV se aplican a pacientes con clínica generalizada. Cada grado se subdivide en grupo A si afecta a las extremidades y como grupo B si la musculatura predominantemente afecta es la bulbar.
- MG-IIA: leve y de predominio en las extremidades
- MG-IIB: leve, de predominio bulbar
- MG-IIIA: moderada, de predominio en las extremidades
- MG-IIIB: moderada, de predominio bulbar
- MG-IVA: grave , de predominio en las extremidades
- MG-IVB: grave, de predominio bulbar
- MG-V: se reserva para los pacientes en crisis miasténica, que necesitan ventilación.
CÓMO SE REALIZA EL DIAGNÓSTICO
El diagnóstico clínico de MG requiere:
- Una historia de debilidad muscular y fatigabilidad que empeoran con el ejercicio.
- La exploración física que demuestre debilidad en distintos grupos musculares y la exclusión de otros diagnósticos.
Exploración neurológica
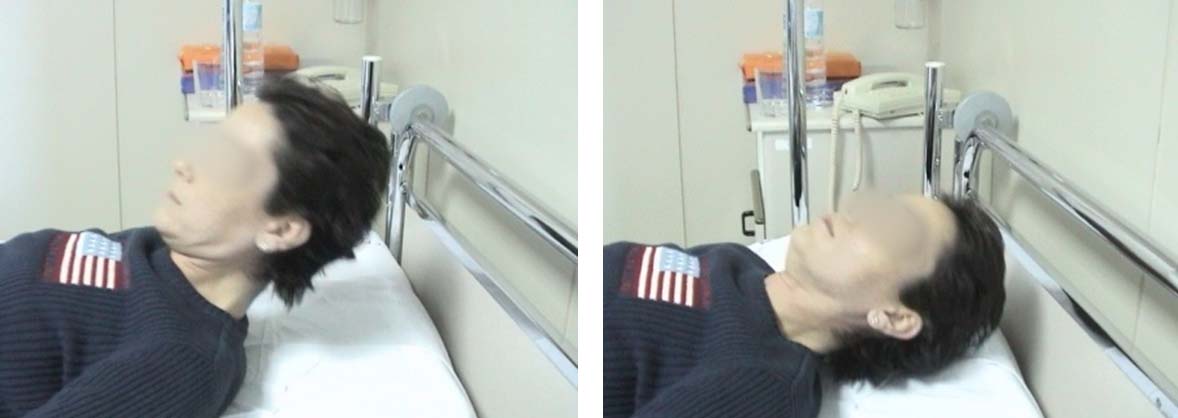
La exploración debe permitir demostrar la existencia de debilidad en distintos grupos musculares al efectuar movimientos repetitivos.
En la imagen de la izquierda se observa como la paciente realiza una flexión normal del cuello y en la imagen de la derecha se objetiva la debilidad de los músculos flexores del cuello al repetir la flexión del mismo.
Ante la sospecha clínica, el paciente debe ser remitido al neurólogo especializado en Miastenia para realizar las siguientes exploraciones complementarias:
Test farmacológico
La administración de cloruro de edrofonio (Tensilón®) (fármaco anticolinesterásico) de uso intravenoso (iv) provoca la mejoría o remisión de los síntomas miasténicos en la mayoría de los pacientes de forma transitoria durante unos miutos. Era la prueba más utilizada para el diagnóstico de M.G hasta la década de los setenta ; pero en la actualidad la más específica es la determinación de los autoanticuerpos.
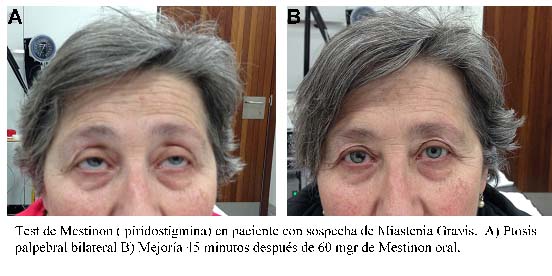
Se puede realizar un test farmacológico con Mestinon oral, en vez de Tensilon, solicitando al paciente que espere en la consulta vunos 30-40 minutos después de ingerir 1 comprimido de Mestinon. Es útil porque además de observar la tolerancia, se puede objetivar con facilidad el beneficio clínico.
Estudios electrofisiológicos
Se utilizan para demostrar la alteración en la transmisión neuro-muscular a nivel como causa de la debilidad de los pacientes. Los test que se utilizan son la estimulación repetitiva que es específica pero puede ser negativa y el estudio de fibra aislada que se reserva para determinados casos, en los que hay dudas diagnósticas porque es de difícil ejecución, nada agradable para el paciente y puede ser positiva en otras patologías.
La estimulación repetitiva se realiza colocando unos electrodos en la superficie de la mano, el hombro o la nariz y registrando en el aparato de EMG el potencial motor obtenido al estimular el nervio de forma repetitiva ( el paciente nota que pasa una corriente eléctrica discretamente molesta).
La fibra aislada se realiza insertando una aguja en un músculo , de la frente o del antebrazo, y una segunda aguja que nuevamente pasa una corriente eléctrica . Es importante la colaboración del paciente para que no haya artefactos en la adquisición de las señales en estas pruebas.

Estudio de autoanticuerpos específicos
- Anticuerpos anti-RACh (Ac.RAch)
Los Ac.RACh son positivos hasta en un 85% de los pacientes con una miastenia generalizada y en un 50-60% de los pacientes con miastenia ocular. Son altamente específicos de MG, aunque se han descrito a títulos bajos en pacientes con timoma, sin clínica de MG y ocasionalmente en otras patologías, inmunomediadas como el lupus y la cirrosis biliar primaria. - Anticuerpos anti-MuSK. (Ac.MuSK)
El 15%-30% de los pacientes con miastenia generalizada en los que no se demuestra la presencia de AcRACh tienen un título positivo de AcMuSK, que en este caso debe ser superior a 0,05nM. La especificidad de la prueba es del 100% y es excepcional encontrar pacientes doblemente positivos a RACh y MuSK. - Anticuerpos anti-LRP4.
No está bien establecido aún el % de pacientes seronegativos para otros anticuerpos que serán positivos para antiLRP4. Se han encontrado positivos en diversas enfermedades inmunomediadas y de momento no son marcadores de un subgrupo de pacientes con miastenia. - Anticuerpos anti cortactina CTTN.
Aproximadamente el 23,7% de pacientes con MG seronegativa y el 9% de los pacientes con MG+RAch tienen estos anticuerpos. Los pacientes tienen una MG ocular o generalizada leve. La determinación de estos anticuerpos es especialmente útil en pacientes con clínica ocular.
Pruebas complementarias
La MG se asocia con otras enfermedades autoinmunes como el hipertiroidismo o el hipotiroidismo. Debe realizarse el estudio de la función tiroidea así como de los anticuerpos antiperoxidasa tiroidea y antitiroglobulina.
Otras enfermedades que se asocian son el vitíligo , el lupus , la artritis reumatoidea y la anemia perniciosa. En estos casos es posible detectar anticuerpos anrtimucosa gástrica
Estudio de imagen del timo
En el abordaje clínico de enfermos con miastenia grave debe descartarse siempre la existencia de timoma. Las técnicas de elección son TC y/o RM torácicas para determinar si hay invasión de estructuras mediastínicas. El porcentaje de MG con timoma es un tema en revisión a nivel internacional en estos momentos. La MG puede ser la primera manifestación del tumor.
DIAGNÓSTICO DIFERENCIAL
El diagnóstico diferencial se debe hacer con:
- Esclerosis lateral amiotrófica
- Miopatías mitocondriales
- Distrofia muscular oculofaríngea
- Miopatía por alteración función tiroidea
- Parálisis agudas motoras, como el botulismo
- Polineuropatía aguda que afecta los pares craneales y la encefalopatía de Wernicke
- Síndrome de Eaton Lambert
- Síndromes miasténicos congénitos
- Estados de fatiga emocional, así como la debilidad muscular histérica o simulada
TRATAMIENTO
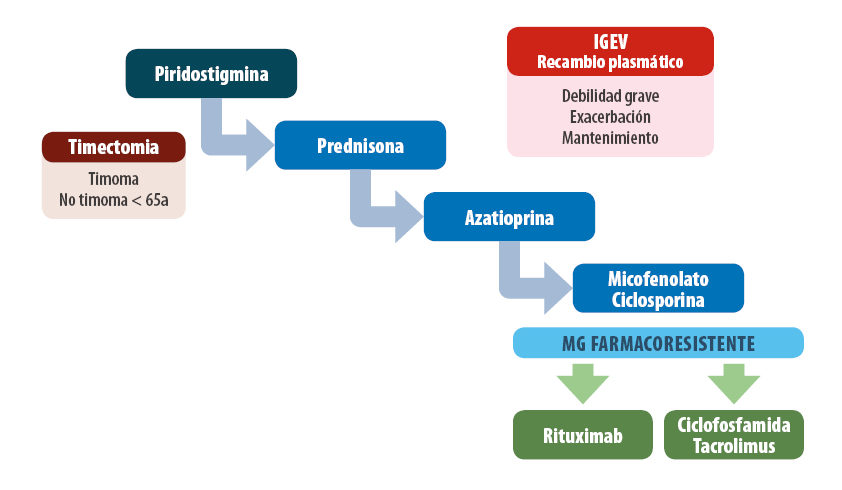
El objetivo del tratamiento es lograr que el paciente esté asintomático o con unos síntomas que le permitan realizar vida relativamente normal con los mínimos efectos secundarios posibles. Este objetivo se logra en más del 80% de los casos.
El tratamiento de los pacientes con MG incluye tres aspectos terapéuticos diferentes:
- Tratamiento sintomático (anticolinesterásicos )
- Timectomía
- Tratamiento inmunológico (inmunosupresores)
- Tratamiento inmunomodulador (plasmaféresis y/o Igev)
Tratamiento sintomático con Anticolinesterásicos
La mayoría de los pacientes MG-RACh+ responden al tratamiento en mayor o menor medida. Sin embargo, menos del 50% de los pacientes MG-MuSK+ responde y de hecho pueden empeorar.
La piridostigmina (Mestinon 60) es el fármaco más utilizado. La dosis inicial oscila entre 120 y 300 mg/día por vía oral repartida en dosis de 30-90 mg. El efecto terapéutico se inicia a los 15-30 minutos , y sus efectos no superan las 4-6 horas. La dosificación es más eficaz si el paciente conoce bien el fármaco y sus efectos y flexibiliza las dosis en función de su estado de fatigabilidad y sus horarios.
Los síntomas secundarios se producen por un exceso de actividad colinérgica, y en la mayoría de las ocasiones son benignos (dolor abdominal, diarrea, calambres y fasciculaciones).
Dos precauciones:
- En general deben trascurrir 4-5 horas entre tomas de Mestinon
- Si un paciente inicia un empeoramiento importante y aumenta significativamente la dosis de Mestinon, puede producirse una crisis colinérgica, que se asocia también con debilidad muscular y se acompaña de sudoración, hipersialorrea, aumento de las secreciones respiratorias, lagrimeo, miosis, nauseas, vómitos y bradicardia.
Timectomía
La timectomía se realizaba en los pacientes con Miastenia desde el año 1934 cuando se publicó por primera vez una mejoría clínica de la enfermedad al operar a un paciente con timoma.
Se considera que la timectomía es beneficiosa en aquellos pacientes con MG RAch+, en pacientes con timoma y en menor grado en pacientes seronegativos.
En 2016 se publicó en New England Journal of Medicine un estudio doble ciego que demuestra que la timectomia juega un papel en el tratamiento de la Miastenia.Así pues, n el momento actual se plantea la timectomía en aquellos pacientes con una MG , con AcRACh+ y una edad comprendida entre los 18 y los 60 años. En los pacientes con timoma, la timectomía se debe realizar independientemente de la edad del paciente.
En los pacientes con una MG-MuSK, las series publicadas no han demostrado efecto beneficioso alguno.
La técnica que se utiliza actualmente es la toracoscopia y solo se realiza la toracotomía si durante la intervención se considera necesaria.
Tratamiento inmunosupresor
Los inmunosupresores han reducido de forma muy importante el número de crisis y la mortalidad por la enfermedad, hay aún un número de pacientes que son resistentes a los fármacos o que éstos les producen unos efectos secundarios importantes, que afectan la calidad de vida de los pacientes.
Hay un consenso internacional que el tratamiento inmunosupresor debe iniciarse con Prednisona. Este fármaco mejora mas del 50% de los pacientes y sus efectos secundarios se pueden minimizar muchísimo si se administra en días alternos y dosis matinal, hecho que permite esta enfermedad. La mejoría clínica se oberva entre las 3 y las 12 semanas.
Después o concomitantemente se aconseja utilizar Aziatoprina o Micofenilato Mofetil con el fin de poder reducir al máximo o suprimir la dosis de prednisona. Estos fármacos pueden ser utilizados como tratamiento único en pacientes con una afectación clínica moderada o leve ya que sus efectos clínicos se notan entre 6 y 12 meses de haber iniciado el tratamiento.
Si hay toxicidad por estos fármacos o los pacientes son resistentes a estos fármacos, o la debilidad muscular requiere un tratamiento a más corto plazo, el neurólogo puede introducir una serie de inmunosupresores, como Ciclosporina, Tacrólimo, Ciclofosfamida o Rituximab.
Es muy importante el seguimiento de los pacientes para detectar cualquiera de los posibles efectos adversos no solo de la prednisona sino de los inmunosupresores (diabetes, hipertensión, osteoporosis, alteración hepática o renal,…).
Tratamiento Inmunomodulador
Existen dos terapias, la Plasmaféresis y las Immunoglobulinas endovenosas que se utilizan cuando los pacientes con MG tienen un empeoramiento clínico importante o están en crisis miasténica porque sus efectos son rápidos pero poco duraderos. Se utiliza también en aquellos pacientes recientemente diagnosticados , con debilidad importante , que necesitan mejorar la debilidad antes que inicien su efecto los fármacos inmunosupresores.
FÁRMACOS QUE PUEDEN AFECTAR A LA MIASTENIA GRAVIS
Se incluye una lista de fármacos, de los cuales prohibidos hay uno (d-penicilamina). El resto de fármacos se pueden utilizar, aunque con la precaución de saber que pueden aumentar la debilidad, de forma que, los pacientes deben advertir al médico que padece MG y los médicos deben advertir a los pacientes que si empeoran han de consultar . Por supuesto que la vigilancia debe ser mayor en aquellos pacientes que ya tienen un cierto grado de debilidad. Se debe consultar de forma urgente con el neurólogo sobre la actitud a tomar, que en algunos casos puede ser la suspensión de la medicación.
Contraindicados:
- D penicilamina
Usar con gran precaución:
- Telitromicina (usar sólo si no hay otra opción posible)
Fármacos que aumentan la debilidad en la mayoría de pacientes:
- Curare y derivados
- Toxina botulínica
- Aminoglucósidos (Gentamicina, Kanamicina, Neomicina, streptomicina, tobramicina)
- Macrólidos (Eritromicina, Azitromicina)
- Fluoroquinolonas (Ciprofloxacino, Levofloxacino, Norfloxacino)
- Quinina, Quinidina y Procainamida
- Interferón-alfa
- Sales de Magnesio (suplementos endovenosos de magnesio)
Fármacos que pueden aumentar la debilidad en algunos pacientes:
- Antagonistas del calcio
- Betabloqueantes
- Litio
- Contrastes yodados
- Estatinas (la relación causal en estos casos puede ser cuestionable debido al uso extendido de estos fármacos)
ANESTESIA GENERAL, ANESTESIA LOCAL, INFILTRACIONES
No hay, en general, más efectos secundarios con anestesia local que los que pueden ocurrir al resto de la población. Lo mismo puede decirse de las infiltraciones con corticoides para problemas locales tipo tendinitis,…
En cuanto a la anestesia general, los pacientes deben advertir al cirujano y sobretodo al anestesista de su enfermedad para que tomen las precauciones necesarias, que en ningún caso debe ser no intervenir al paciente. La anestesia general debe ser administrada en un centro que disponga de una Unidad de Vigilancia postquirúrgica o una UCI por si el paciente tarda más de lo habitual en poder ser desintubado.
ALIMENTOS PROHIBIDOS
No hay ningún estudio que muestre que algunos alimentos empeoran la situación clínica de los pacientes, excepto aquellos que incluyan compuestos que ya están listados entre los fármacos, como puede ser el agua tónica.
VACUNA DE LA GRIPE
Hay una controversia sobre la utilización de estas vacunas. Aunque hay pacientes que han empeorado su clínica, parece que no ocurre en la mayoría de los casos. El riesgo se considera mayor si la vacunación se realiza con microorganismos atenuados (vivos) y menor con microorganismos inactivados (muertos). Por eso, se recomienda que el médico decida si el paciente se beneficiaría de la vacuna por estar inmunodeprimido, edad,… y si se produce un empeoramiento clínico que lo remita al neurólogo responsable.
EMBARAZO
La Miastenia inmunomediada no es hereditaria. Puede ocurrir que durante el primer trimestre del embarazo haya un empeoramiento clínico, pero en general durante todo el embarazo los pacientes están estables pudiendo ser tratados, si lo necesitan con algunos de los fármacos inmunosupresores mencionados.