General information
Myasthenia gravis (MG) is a rare, chronic neuromuscular disease characterized by fatigue and fluctuating weakness of the voluntary muscles. Weakness can affect any muscle group, but it most commonly affects ocular muscles, limbs, or muscles responsible for respiration, swallowing and phonation.
The disease is autoimmune – a condition where the body’s own immune system mistakenly attacks its own tissues. Antibodies that are normally produced by the body to fight against foreign proteins mistakenly target the body’s own proteins. These malfunctioning antibodies are named autoantibodies.
MG is treatable so accurate diagnosis is key.
Ideally, patients should be treated by expert neurologists who work in close communication with the patients and their family physicians.
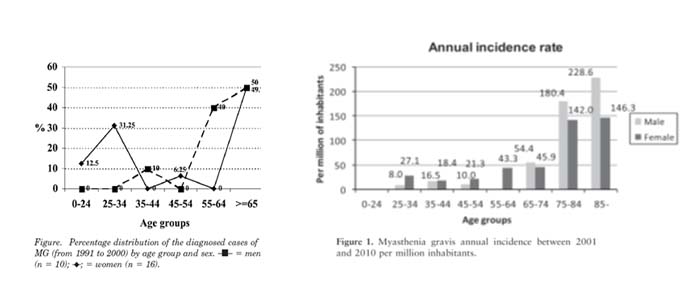
Is MG A DISEASE OF THE YOUNG?
MG was initially considered to predominantly affect individuals aged 15 and 35 years, particularly women. However, according to recent studies in several countries, it has been found that MG develops after the age of 60 in over 65% of cases. Therefore, because response to treatment is good – particularly in the elderly – when clinical signs are compatible with MG, the disease should be included in the differential diagnosis, regardless of age.
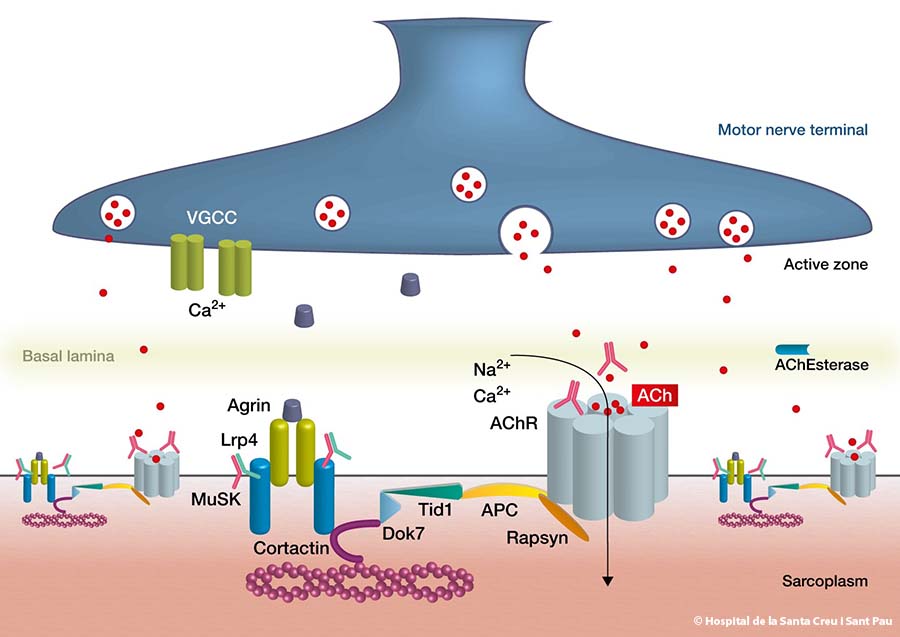
WHAT DO WE KNOW ABOUT THE AUTOANTIBODIES IN MG?
It is not yet clear what triggers the autoantibodies in MG, but we know that they impair communication between nerves and muscles. Four specific autoantibodies have been identified to date at the neuromuscular junction. The majority of patients with MG have one of these four autoantibodies
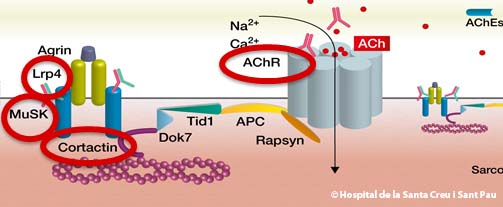
- Over 80% of patients have autoantibodies against the acetylcholine receptor located in the post-synaptic membrane of the neuromuscular junction. When this autoantibody is identified in a blood sample, the MG is defined as RAch-MG
- Some patients (5-15%) who are MG-RAch negative have autoantibodies against a molecule known as MuSK, which is also located in the post-synaptic region of the neuromuscular junction and is defined as MuSK-MG
- A third autoantibody has been identified in a small number of patients. Recent studies suggest 1 to 3% of all patients with myasthenia gravis have these antibodies, defined as LRP4-MG
- A fourth autoantibody has been described against cortactin (CTTN). It has been found in around 25% of patients with seronegative MG and 9% of patients with MG + AchRAb. It is not associated to MuSK and LRP4 MG. This subtype of MG is defined as CTTN-MG.
Other autoantibodies against other molecules, such as agrin and collagen Q, have been described in patients with AChR-MG, MUSK-MG, or LRP4-MG.
The 5-7% of patients in whom none of the above autoantibodies have been demonstrated are considered “seronegative MG”. These patients, however, also respond to immunosuppressive treatments.
CLINICAL MANIFESTATIONS
As stated above, the main clinical symptom of MG is muscle fatigue accompanied by muscle weakness. This fatigue worsens throughout the day or with repeated muscle activity, and improves partially or totally with rest. The signs and symptoms of myasthenia are directly related to the muscles involved:
- In 85-90% of patients, the first symptom of MG is weakness of the ocular muscles, causing diplopia (double vision) and unilateral or bilateral ptosis (pathological droopy eyelid). These symptoms vary throughout the day and may not always be present.
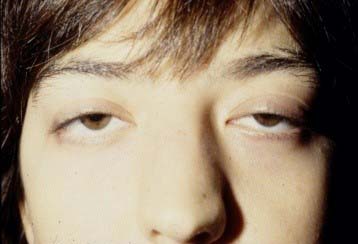
Bilateral Ptosis - Another common symptom is weakness of facial muscles, especially of the orbicularis oculi and lips.
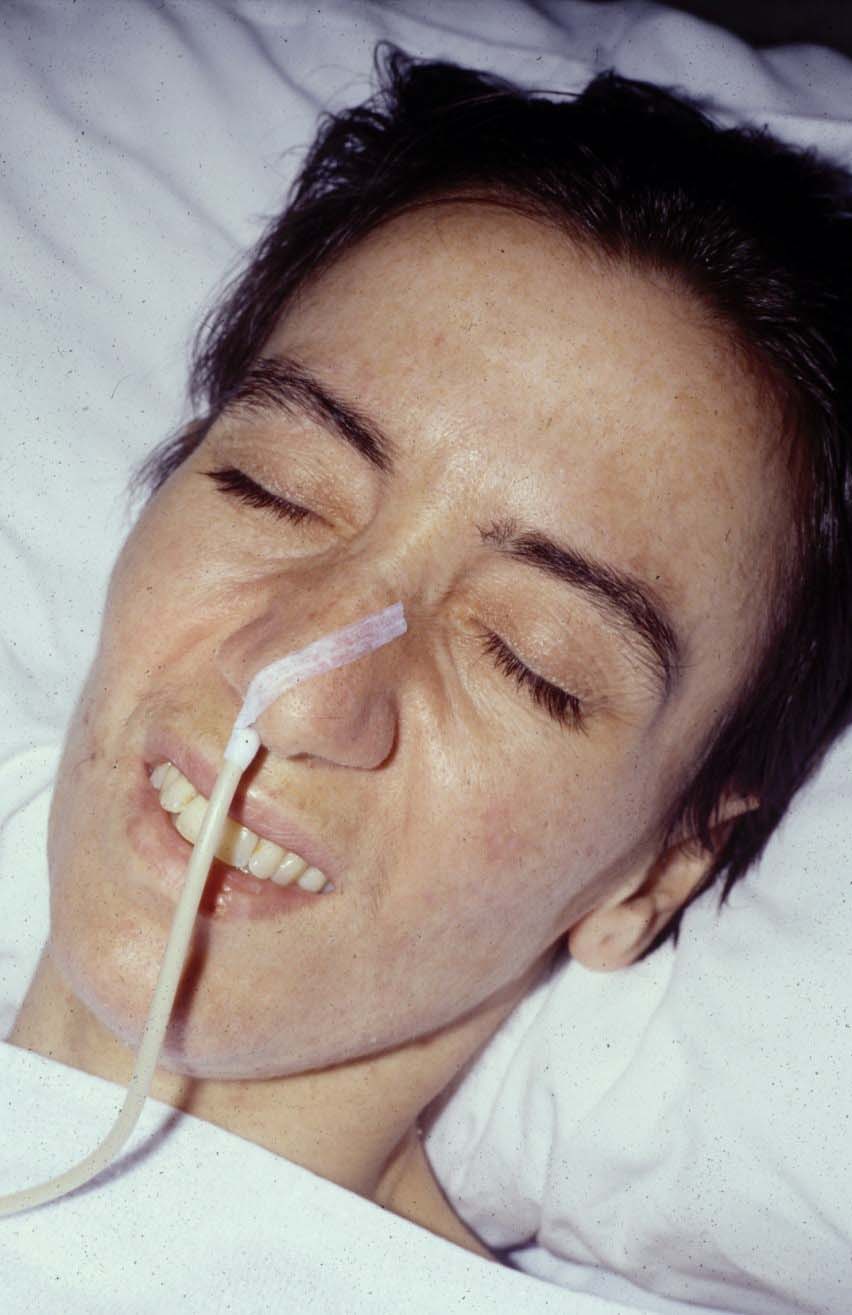
Orbicularis oculi and orbicularis oris - The cervical muscles are also commonly involved, causing weakness in flexion/extension of the neck
- The masseter muscles may also be affected, causing jaw claudication (pain on chewing)
- Patients also frequently complain of weakness and fatigability of arm and leg muscles, especially at the proximal level, although they may also report weakness of distal muscles
- In elderly patients or patients with MuSK+MG, the bulbar muscles are often involved, and can even be the first signs of the disease. Bulbar involvement causes dysphagia, dysarthria and dyspnea. These symptoms must be recognized as early as possible because they can be life-threatening.
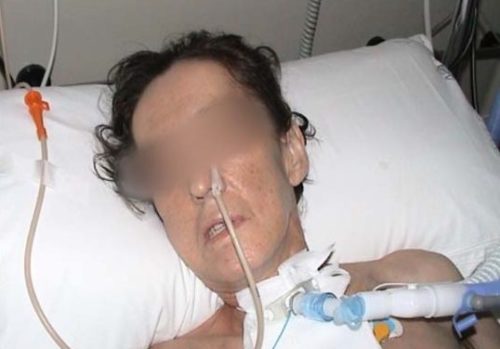
- The respiratory muscles may also be affected, and in such cases a myasthenic crisis can occur. In this event, hospital admission is required and mechanical ventilation may be needed for acute respiratory failure
CLINICAL CLASSIFICATION
In 2000, the Myasthenia Gravis Foundation of America (MGFA) proposed the classification that is most commonly used today.
Ocular MG:
- Class I: Any ocular muscle weakness; may have weakness of eye closure. All other muscle strength is normal.
Generalized MG:
- Class II: Mild weakness affecting muscles other than ocular muscles; may also have ocular muscle weakness of any severity.
- IIa. Predominantly affecting limb, axial muscles, or both. May also have lesser involvement of oropharyngeal muscles.
- IIb. Predominantly affecting oropharyngeal, respiratory muscles, or both. May also have lesser or equal involvement of limb, axial muscles, or both.
- Class III: Moderate weakness affecting muscles other than ocular muscles; may also have ocular muscle weakness of any severity.
- IIIa. Predominantly affecting limb, axial muscles, or both. May also have lesser involvement of oropharyngeal muscles.
- IIIb. Predominantly affecting oropharyngeal, respiratory muscles, or both. May also have lesser or equal involvement of limb, axial muscles, or both.
- Class IV: Severe weakness affecting muscles other than ocular muscles; may also have ocularmuscle weakness of any severity.
- IVa. Predominantly affecting limb, axial muscles, or both. May also have lesser involvement of oropharyngeal muscles.
- IVb. Predominantly affecting oropharyngeal, respiratory muscles, or both. May also have lesser or equal involvement of limb, axial muscles, or both.
- Class V: Defined as intubation, with or without mechanical ventilation, except when employed during routine postoperative management. The use of a feeding tube without intubation places the patient in class IVb.
Diagnosis of MG
The clinical diagnosis of MG requires:
- A history of muscle weakness and fatigue that worsens with exercise.
- A physical examination showing weakness in some muscle groups
- Exclusion of other diagnoses.
Neurological examination
The examination addresses weakness of muscle groups during exercise.
The image below shows weakness of neck flexor muscles (right) after a normal neck flexion (left)
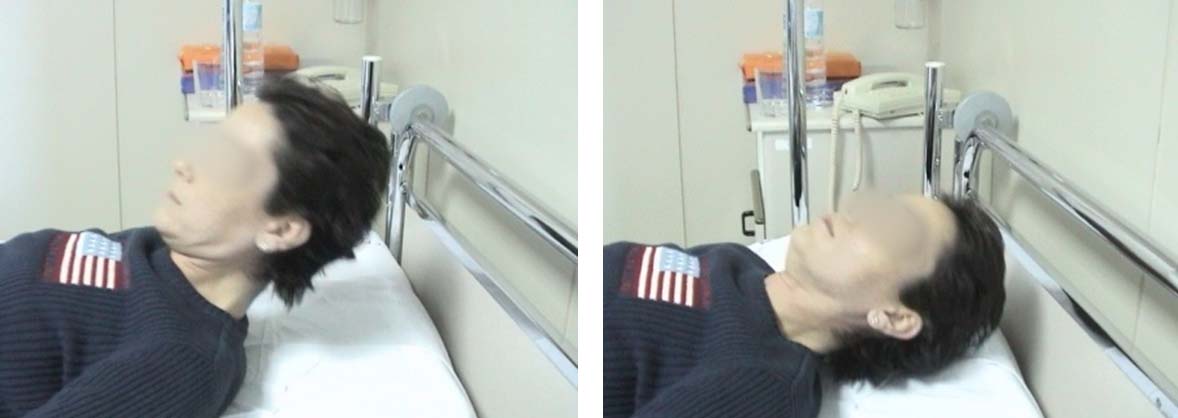
Fatigability of neck flexors
MG should be considered in the diagnosis in patients who report diplopia and ptosis, and also in patients – especially the very elderly – who have dysarthria or dysphagia that varies throughout the day. It is not uncommon that these patients are misdiagnosed as having some more prevalent disease, such as cerebral vascular pathology, or even a psychological disorder.
Several tests may be conducted for the diagnosis of MG:
Pharmacological tests
Until recently, the most common pharmacological test in the diagnosis of MG was the intravenous (IV) administration of edrophonium chloride (Tensilon®) (an anticholinesterase drug). In most patients, this drug improves myasthenia symptoms, such as diplopia or ptosis, for a few minutes after injection by increasing the muscarinic effects of acetylcholine. It should be used cautiously, however, especially in older adults and those with cardiac disease or bronchial asthma.
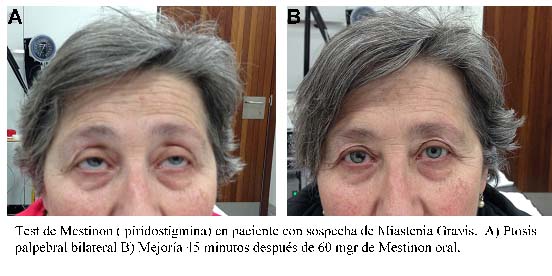
An alternative pharmacological test may be performed using oral Mestinon (pyridostigmine). For this test, the patient must remain in the office for 30-40 minutes after taking the drug so as to evaluate the clinical benefit (see figure) and tolerance to the drug.
.
Electrophysiological studies
Electrophysiological studies are used to determine alterations in neuromuscular transmission. Two tests are commonly used. The first of these is repetitive nerve stimulation. This test is specific but has the drawback that it may produce false negative results. The second option is single fiber electromyography (SFEMG). This test is more time-consuming and it can be positive for other diseases.
Repetitive nerve stimulation is performed by placing surface electrodes over the endplate region on the hand, shoulder, or nose. The motor potential is then determined by repetitively stimulating the motor nerve (the patient feels a small electric current). In MG, the amplitude of the motor potential shows a progressive decline after the first four to five stimuli (decremental response).
For the SFEMG test, a needle is inserted into a muscle in the forehead or the forearm, and a second needle passes an electric current. This technique records the action potentials of two muscle fibers innervated by the same motor axon. The variability in time of the second action potential in relation to the first is called “jitter.” This technique is positive in more than 90% of patients with MG if a facial muscle is studied. As artifacts may disturb the recordings, the patient’s collaboration is essential.

Blood tests for the specific autoantibodies
Anti-AChR antibodies (AchR Ab)
Anti-AChr autoantibodies are determined using RIA (radioimmunoassay). Although these antibodies are highly specific for MG, titers may occasionally be positive in patients with thymoma and no clinical signs of MG, and in patients with other immune-mediated disorders such as lupus or primary biliary cirrhosis.
Anti-MuSK antibodies (MuSK Ab)
Anti-MuSK autoantibodies are determined using RIA (radioimmunoassay). The test for these antibodies is specific but a few patients may be doubly positive for AChR and MuSK or LRP4 and MuSK.
Anti-LRP4 antibodies
Autoantibody detection is performed using transfected cells. Patients with these antibodies tend to have mild-to-moderate symptoms. Some patients have both anti-LRP4 antibodies and MuSK antibodies.
CTTN anti-cortactin antibodies
To determine anti-cortactin antibodies the Immunoblot and ELISA test with recombinant Cortactin are used. These patients have ocular or mild generalized MG. The study of these antibodies is especially useful in patients with ocular symptoms.
Coexisting disorders
MG can be associated with other autoimmune diseases such as hyperthyroidism or hypothyroidism. It may also be associated with vitiligo, lupus, rheumatoid arthritis, or pernicious anemia.
Thymus pathology
Thymus pathology, both hyperplasia and thymoma, should be ruled out. The techniques of choice here are CT and/or thoracic MRI to determine the size of the thymus and, in thymomas, to assess invasion of mediastinal structures. Titin and Ryanodine receptor antibodies are often present in patients with thymoma.
TREATMENT
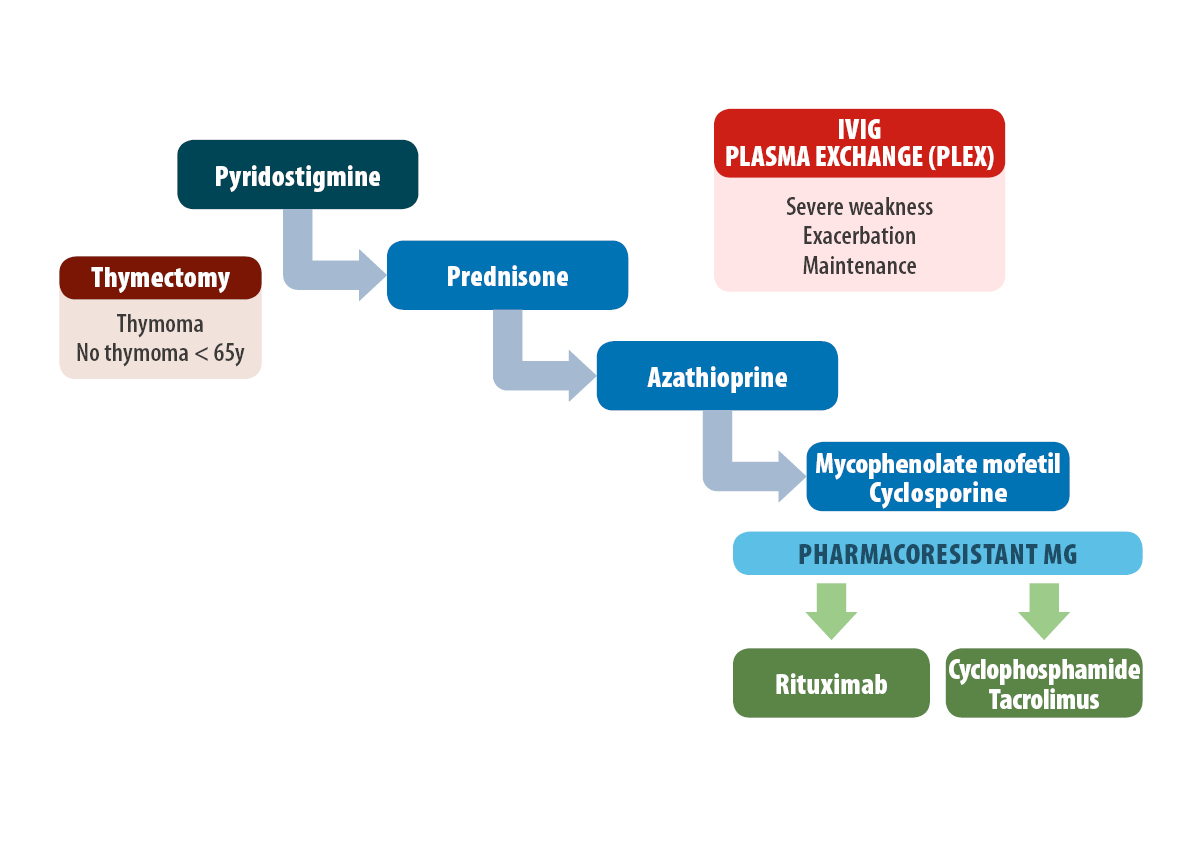 The goal of treatment for MG is to manage symptoms or achieve clinical remission with minimal medication and side effects. Patients achieve a good quality of life in more than 80% of cases.
The goal of treatment for MG is to manage symptoms or achieve clinical remission with minimal medication and side effects. Patients achieve a good quality of life in more than 80% of cases.
Treatment of MG includes several therapeutic aspects:
Symptomatic therapy (acetylcholinesterase inhibitors)
Thymectomy
Immunosuppressive drugs
Immunomodulatory treatment (plasmapheresis or IVIG)
Symptomatic therapy (acetylcholinesterase inhibitors)
Pyridostigmine is the most commonly used acetylcholinesterase inhibitor drug for the treatment of symptoms. Most AChR+MG patients respond to treatment to some degree. However, less than 50% of MuSK + MG patients respond, and the disorder may even progress in some cases.
The initial dose of pyridostigmine ranges from 120 to 300 mg / day orally, divided into doses of 30-90 mg. The therapeutic effect appears in 15-30 minutes and lasts 4-6 hours. Dosing is more effective if the patients know the drug and its effects well and adjust their daily dose in function of their fatigability status and their schedules. The effect of pyridostigmine remains unchanged over time.
The side effects are caused by an excess of cholinergic activity and they are benign in most cases (abdominal pain, diarrhea, cramps and fasciculations).
Too high a dose may trigger a cholinergic crisis. Such an event is associated with muscle weakness and is accompanied by sweating, hypersalivation, increased respiratory secretions, lacrimation, miosis, nausea, vomiting, and bradycardia.
.
Thymectomy
In patients with thymoma, a thymectomy should be performed independently of the patient’s age.
In 2016, an international, randomized, controlled trial published in the New England Journal of Medicine confirmed that thymectomy plays a role in the treatment of myasthenia. The study showed a benefit in patients with generalized AChR +MG, a disease duration of less than 3 to 5 years, and age between 18 and 60-65 years.
The preferred technique for thymectomy is currently the video- and robot-assisted approach. Traditional transsternal thymectomy is only performed if the surgeon considers this technique is necessary.
A thymectomy is not usually recommended for patients with ocular or seronegative MG although it may be considered when other drug treatments fail.
In patients with a MuSK+MG, thymectomy does not appear to show any beneficial effects.
Immunosuppressive drugs (IS)
Several guidelines for MG support the use of immunosuppressive drugs.
An international consensus states that IS treatment should start with prednisone/ prednisolone. This drug improves the disorder in more than 50% of patients and its side effects can be greatly minimized if it is administered in morning doses on alternate days. A clinical improvement is usually observed after 4 to 12 weeks.
The concomitant use of azathioprine or mycophenolate mofetil is advisable to reduce or suppress the dose of prednisone in the long term. These drugs can also be used as a single treatment in patients with mild clinical involvement because their clinical effects occur between 6 and 12 months after starting treatment.
In the case of severe side effects, patient resistance to these drugs, or muscle weakness requiring further treatment, the neurologist can introduce a series of IS such as cyclosporine, tacrolimus, cyclophosphamide, or rituximab.
IS have greatly reduced not only the number of patients who require intubation but also mortality due to the disease. However, new therapeutic approaches are still needed to improve the quality of life in drug-resistant patients and in those who have severe side effects..
Immunomodulator treatment
Plasma exchange (PLEX) and intravenous immunoglobulins (IVIG) may be used when patients with MG present significant clinical worsening with life-threatening signs such as respiratory insufficiency or dysphagia. PLEX or IVIG may also be used when a rapid response is needed in recently diagnosed patients with severe weakness, when weakness must be improved before a thymectomy can be performed, and before immunosuppressive drugs start their effect.
IVIG, and more recently, subcutaneous immunoglobulins (SCIG) may be used as “maintenance therapy” for patients with refractory MG.
DRUGS THAT CAN AFFECT MYASTHENIA GRAVIS
Although only one drug – D-penicillamine – is contraindicated in patients with MG, a considerable number of drugs have been reported to exacerbate weakness. Many of these appear to aggravate weakness in the majority of patients, but others induce only a slight degree of weakness and affect only a few patients. Physicians should give patients a list of medications that might exacerbate their symptoms.